




We work with our customers in precision medicine to solve problems in engineering, bioinformatics, science, and translational medicine that involve clinical and omics data.
We work with our customers in precision medicine to solve problems in engineering, bioinformatics, science, and translational medicine that involve clinical and omics data.
Genomics Shaping Healthcare
Genomics Shaping Healthcare



Deep First Hand Experience
We have deep first-hand experience in the area, spanning tens of thousands of reports.

Technology
Our fast and nearly automated platform for small and large oncology panel reporting is based on ~10 years curation and automation.

Customer Experience
Our teams have helped build germline and somatic variant interpretation tools for multiple US diagnostic and device companies.


Data Generation
We generate single-cell, spatial transcriptomics and genomics data from clinical samples in our India-based CAP lab.

Data Curation
We curate publicly available and client-specific omics data and develop custom ontologies for metadata and cell type annotations, with focus on single-cell and spatial transcriptomics.

Data Analysis
We build bioinformatics and tertiary analysis workflows that help go from raw reads to insights, and package these in software platforms to help lab scientists, bioinformaticians, and curation scientists.


Track Record
We have a 20-year strong track record in product development for omics applications, e.g., GeneSpring, MassProfiler Professional, and StrandNGS.

Wide Range of Experience
We have experience building visualization tools, rules engines, data management systems, user interfaces, and algorithms. We have built applications for microarrays, genomics, transcriptomics, metabolomics, and proteomics, as well as for metadata management, clinical reporting, electronic data capture, and provider portals.

Software Development for
Regulatory Compliance
We develop software for HIPAA- and FDA-compliant software accompanying medical devices and genomics tests.
Excellence Through the Years
Excellence Through the Years
Drawing on 25+ years of domain experience, we are a team of over 300 highly trained curation scientists, software engineers, testing engineers, bioinformaticians, and clinical researchers.
We are headquartered in Bangalore, where we were founded, and maintain close ties with the Bay Area and San Diego, where many of our customers are based.

Quality
Quality is a core tenet of everything we do at Strand. Our team of over 500 ensures that every deliverable meets the highest standards through rigorous internal checks and validation.

Innovation
Our team of over 340 specialists continuously adapts to and innovates on breakthroughs in the ever-evolving field of precision omics, leveraging cutting-edge technologies such as AI-based solutions, real-world data (RWD) insights, and multiomics analysis.

Collaboration
We work closely with clients across academic, pharmaceutical, bioinformatics, biotechnology, and software sectors, providing solutions in data harmonization, precision analytics, software and platform development, and quality control, among others.

Global Perspective
Compliant with internationally recognized standards such as HIPAA and ISO 27001, and backed by CAP accreditation for our NGS laboratory, our global team serves clients worldwide.

Testimonials



I worked with Strand on maintaining and enhancing two critical systems. The work encompassed multiple technology areas, requiring diverse technical skills. They were quick learners, taking on new projects with enthusiasm and delivering highly polished, visually appealing reports crafted to exacting standards. I was impressed with the offshore team's attention to detail and timely delivery of high-quality results.
Lead Software Engineer
Early Stage Multi-Cancer Detection Company, California

Strand did a fantastic job helping us curate a canine database for the purpose of assay design, interpretation, and reporting. They worked as an extension of our team and played a critical part in helping us with bioinformatics and laboratory protocols that supported the development of the oncology test.
Chief Technology Officer
a canine diagnostics company

We were very impressed with the quality of work and timeliness; you’re definitely our go-to for bioinformatics consulting.
Director, Bioinformatics
Illumina

We were immensely impressed by Strand's ability to rapidly recruit a substantially sized clinical cohort of cancer patients, and to design and run a complex liquid biopsy panel on samples drawn from the cohort, all in roughly a year's time.
Dr. Nishant Agarwal
Chief of Otolaryngology-Head and Neck Surgery and Director of Head and Neck Surgical Oncology, University of Chicago

Strand did a fantastic job helping us characterize the analytical performance of our gene expression panel. They worked as an extension of our team, and played a critical part in ramping up our assay development efforts.
Vice President - Research and Development
Early Stage Molecular Diagnostic Company, San Francisco

We have been using the StrandOmics pipeline to analyze and generate a report for our clinical cancer panels for over three years now. I would highly recommend using it to analyze data generated from clinical cancer NGS panels and the outputted clinical report provided after analysis.
Senior Scientist/Medical Laboratory Director for NY State
PrimBio Research Institute

Prathyush [Strand developer] has been my go-to person for complex stories. He has always had a holistic approach to the solution instead of diving right into the problem
Engineer
Illumina

09 Mar 2026
Written by Suhasini Singh

19 Jan 2026
Written by Chinta Sidharthan (Strand Life Sciences) and Devika Garg (TileDB)

15 Dec 2025
Written by Suhasini Singh
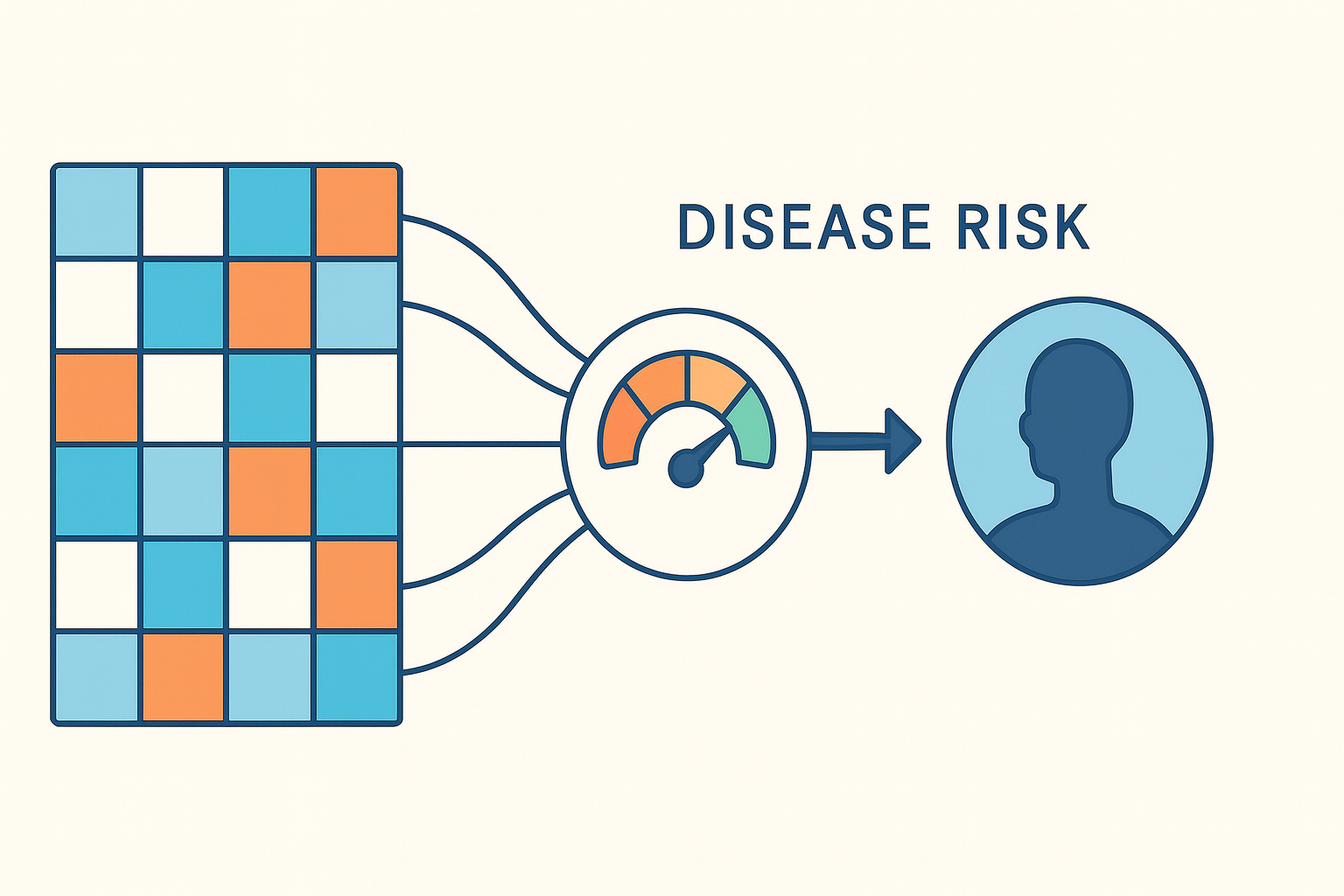
26 Nov 2025
Written by Sharon Christella

17 Nov 2025
Written by Sharon Christella
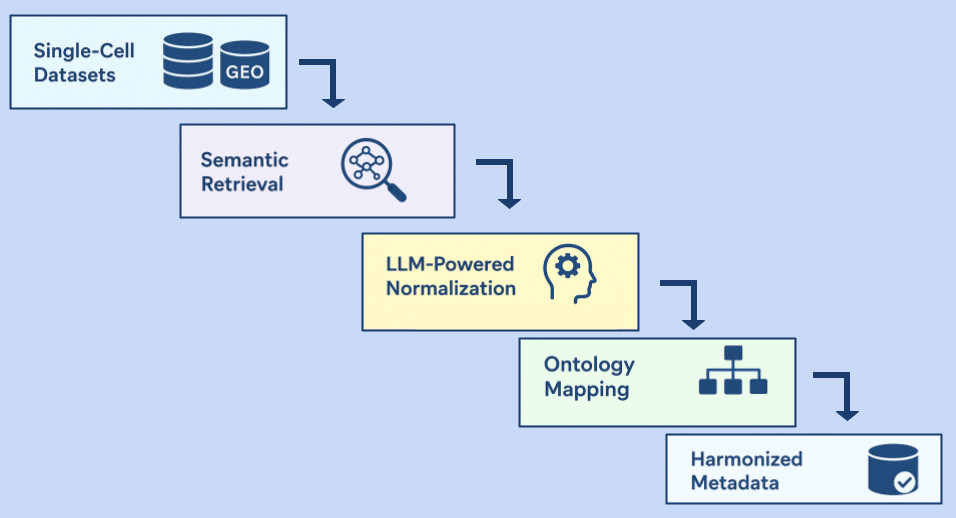
11 Sep 2025
Written by Badri Padhukasahasram, Shrutee Jakhanwal and Chinta Sidharthan
Insights from our CEO
Insights from our CEO




Leaders in Genomics




