Tumor-normal matched sequencing is a key advancement in liquid biopsy, as it enhances/ improves the specificity of somatic mutation detection in circulating tumor DNA (ctDNA).
What is tumor-normal sequencing, and how is it applied to liquid biopsies?
Tumor–normal sequencing, or matched sequencing, involves analyzing both a patient’s tumor DNA and their “normal” DNA. The “normal” DNA is sourced either from adjacent normal tissues or from white blood cells (WBCs). This form of sequencing allows:

This reduces false positives and ensures that only true tumor-specific mutations are reported.
When applied to liquid biopsies, tumor-normal sequencing is adapted to circulating cell‑free DNA (cfDNA). A blood draw yields both cfDNA (which contains fragments of tumor DNA shed into the bloodstream) and genomic DNA (from WBCs). Sequencing both fractions at high depth, then filtering out any variants present in the WBC DNA, enables the precise reporting of somatic mutations.
This approach dramatically improves the specificity of liquid biopsy assays by minimizing false positives and enhancing confidence in minimal residual disease monitoring, treatment resistance detection, and biomarker assessment.
When can tumor-matched sequencing using liquid biopsies be used?
Tumor-matched sequencing for liquid biopsies is particularly useful when:
- Tumor tissue is unavailable: Biopsy may be risky or not feasible due to location, patient condition, or ethical concerns for rebiopsies.
- To eliminate false positives and avoid unnecessary/misguided therapies: Studies have shown that tumor-only sequencing can misidentify germline or CHIP variants as somatic, especially in genes like TP53 and DNMT3A. Brannon et al. (2021) showed that in a clinical assessment of 681 blood samples, tumor-matched sequencing could detect somatic alterations in 73% of the cases and identified that 56% of these alterations were clinically actionable variants. Critically, the study showed that this method could filter out over 10,000 germline or CHIP variants, which dramatically reduced the numbers of false positives. Misannotating germline variants as somatic can lead to unnecessary and potentially harmful treatments. Similarly, Ptashkin et al. (2018) showed that across 17,469 advanced cancer patients, 5.2% would have harbored CHIP variants falsely designated as tumor mutations, nearly half of which were “oncogenic” variants that could prompt erroneous therapeutic decisions.
- Accurate detection of minimal residual disease (MRD): Tracking residual disease post-treatment demands absolute accuracy. Tumor-matched sequencing using blood samples allows for high confidence somatic calls that enables tracking of MRD and early relapse detection, often before radiologic changes occur, enabling timely interventions.
- Serial monitoring to guide therapeutic insights: Serial tumor-normal ctDNA analysis empowers clinicians to monitor emerging resistance mutations (e.g., KRAS, TP53) in real time, guiding adaptive therapy strategies and enrollment in precision clinical trials. For example, Frydendahl et al. (2024) showed that tumor-normal sequencing analysis of serial blood samples could detect recurrence in patients at a median of 8.7 months before imaging.
- Reliable biomarker assessment: Calculating metrics like tumor mutational burden (TMB) or microsatellite instability depends on accurate somatic calls. Germline contamination can distort these measurements, compromise immunotherapy decisions, and hinder biomarker-driven treatment protocols.
How is tumor-matched sequencing performed in blood samples?
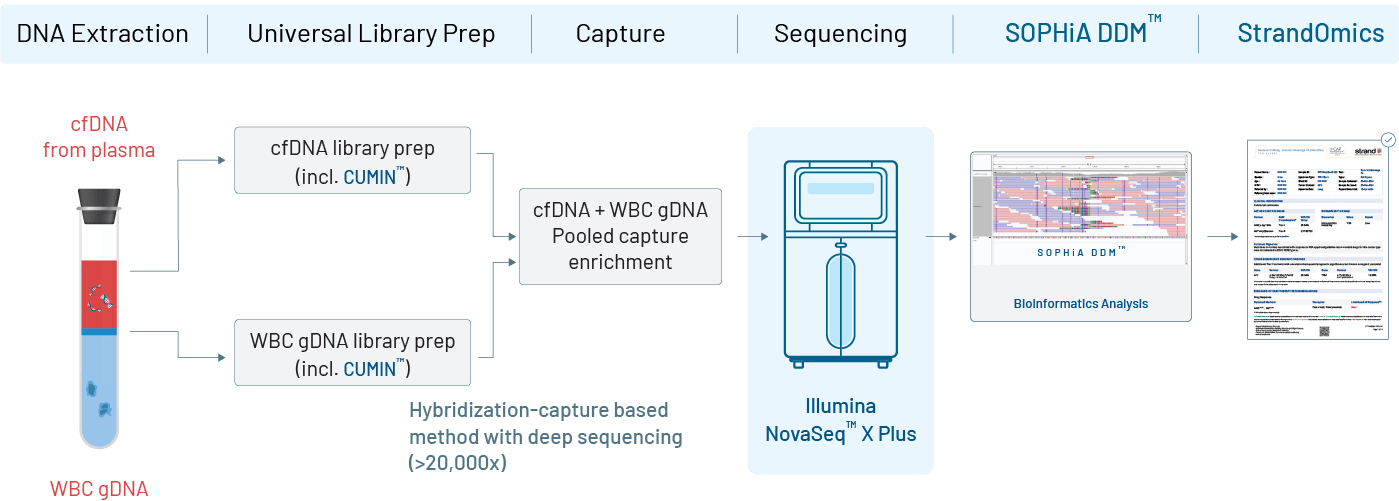
In a tumor–normal matched liquid biopsy, a single blood sample is used to isolate circulating tumor DNA (ctDNA) from plasma and genomic DNA from white blood cells (WBCs) as the matched normal reference. Both DNA types are processed using advanced library preparation techniques, with ctDNA tagged by unique molecular identifiers (UMIs) to improve accuracy. The ctDNA is sequenced at very high depth (~20,000×) to detect low-frequency tumor variants, while WBC DNA is sequenced at lower depth (~1,000×) to help identify non-tumor mutations. Bioinformatic analysis uses the WBC data to filter out germline variants and clonal hematopoiesis–related changes, ensuring only true tumor-specific mutations are reported. The final report highlights clinically relevant, actionable variants to guide diagnosis, treatment decisions, and disease monitoring.
Strand’s offerings for tumor-normal matched sequencing for liquid biopsies
Strand Life Sciences offers MSK-ACCESS® (Memorial Sloan Kettering Cancer Center – Analysis of Circulating cfDNA to Evaluate Somatic Status) in India, a highly sensitive assay (developed using data from >25,000 tumors) that uses hybridization capture, molecular barcoding, and ultra-deep sequencing on cell-free DNA samples to:
- Identify low-frequency genomic alterations
- Detect rare mutations in cell-free DNA
The test identifies genomic alterations such as:
- Single-nucleotide substitutions
- Small insertions and deletions
- Copy number alterations
- Selected rearrangements
- Microsatellite instability
We integrate MSK-ACCESS® with SOPHiA DDM™ for reliable NGS analysis and the expertly curated knowledgebase of StrandOmics for comprehensive clinical interpretation. Currently, MSK-ACCESS® assays for these alterations in 148 key cancer-associated genes that can be targeted with either an approved or investigational drug.
Overall, our services empower healthcare providers to monitor disease progression, detect recurrence, and tailor treatment strategies for optimal patient outcomes in cancer.
References:
- Mandelker D, Ceyhan-Birsoy O. Evolving significance of tumor-normal sequencing in cancer care. Trends in cancer. 2020 Jan 1;6(1):31-9.
- Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, Meng F, Haque M, Sadowska J, Murphy BJ, Baldi T. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nature communications. 2021 Jun 18;12(1):3770.
- Ptashkin RN, Mandelker DL, Coombs CC, Bolton K, Yelskaya Z, Hyman DM, Solit DB, Baselga J, Arcila ME, Ladanyi M, Zhang L. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA oncology. 2018 Nov 1;4(11):1589-93.
- Matched tumor-normal sequencing: The preferred method for identifying somatic mutations driving tumorigenesis – SOPHiA GENETICS. https://www.sophiagenetics.com/resource/matched-tumor-normal-sequencing-preferred-method-identifying-somatic-mutations-driving-tumorigenesis/; First accessed 11th july 2025
- Finkle JD, Boulos H, Driessen TM, Lo C, Blidner RA, Hafez A, Khan AA, Lozac’hmeur A, McKinnon KE, Perera J, Zhu W. Validation of a liquid biopsy assay with molecular and clinical profiling of circulating tumor DNA. NPJ Precision Oncology. 2021 Jul 2;5(1):63.
- Liu L, Tsai J, Luong K, Bilke S, Davila M, Ding Y, Do MV, Gibson J, Movsesyan A, Roessler K, Song F. A highly sensitive, low input, and automated assay for molecular residual disease detection (MRD) using whole-genome sequencing (WGS).
- Frydendahl A, Nors J, Rasmussen MH, Henriksen TV, Nesic M, Reinert T, Afterman D, Lauterman T, Kuzman M, Gonzalez S, Glavas D. Detection of circulating tumor DNA by tumor-informed whole-genome sequencing enables prediction of recurrence in stage III colorectal cancer patients. European Journal of Cancer. 2024 Nov 1;211:114314.